
 circlize绘制基因组圈图的原理与使用
circlize绘制基因组圈图的原理与使用
由于图形大多由点、线、多边形等简单图形组成,circlize实现了在圆形作图区域添加图形的低级绘图函数,通过低级绘图函数的不同组合,可以轻松生成更复杂的图形。这一原则确保了各种类型的高级图形不会受软件包本身的通用性限制,并且可以在此基础上构建针对特定兴趣的高级包。
# circlize的原理
circlize会把一块画布上的常见坐标系统转化成极坐标系统,并将其标注成普通的二维坐标系。这一块区域就被称为track,不同track组成了一个形如圈的sector。 ![]()
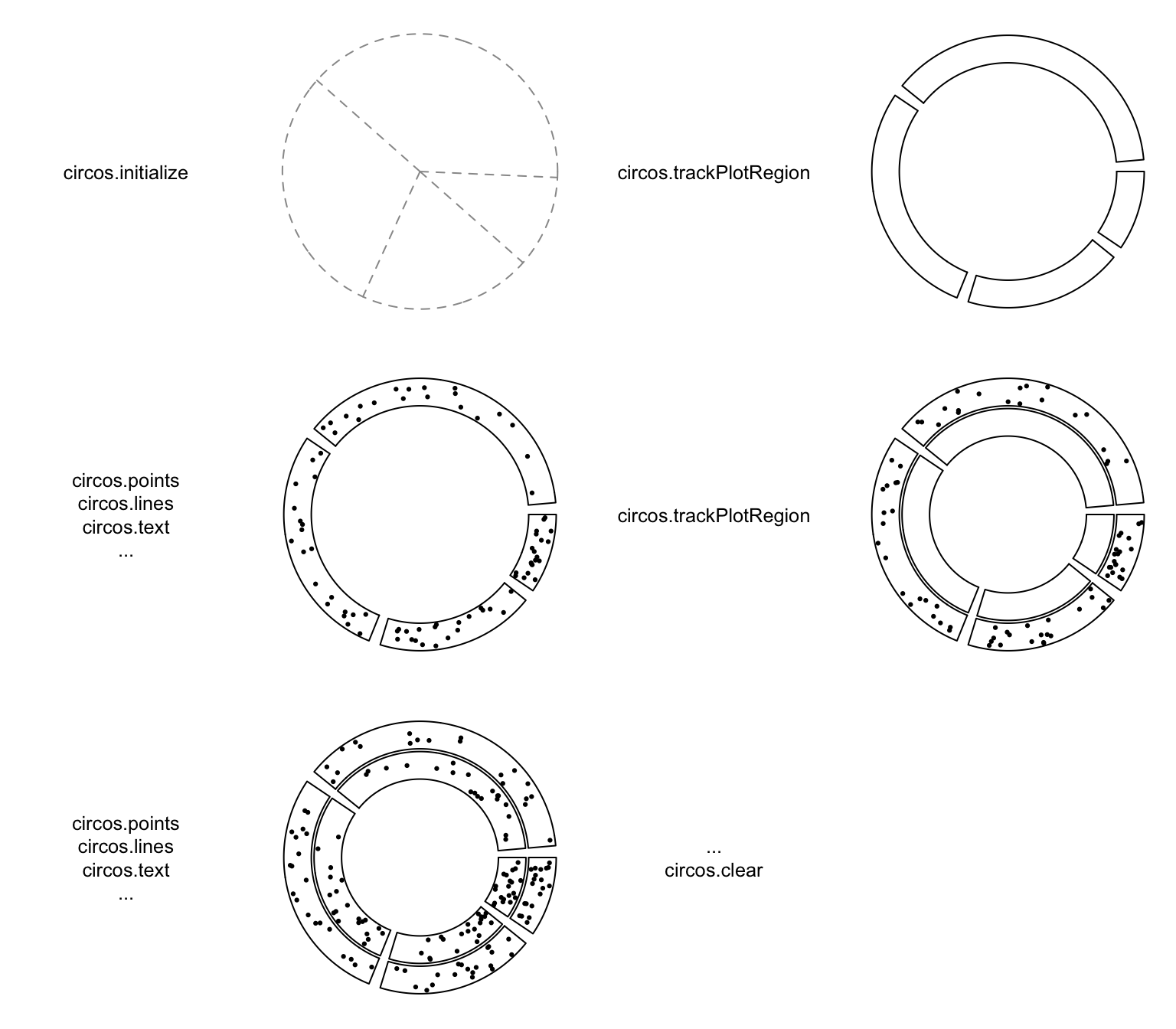
# panel.fun参数
panel.fun通常会在当前track区域进行绘制,为了能够批量在当前sector绘制图形,可以设置sector.index=get.cell.meta.data(sector.index)以及track.index=get.cell.meta.data(track.index),通过trackPlotRegion来绘制新的sector。panel.fun的通常设置形式是panel.fun=function(region,value,...){...}。circos.genomicTrackPlotRegion接受的输入形式为数据框,这可以类比为一个bed文件。一个通常的bed文件应该至少有四行,region就是前三列,表示了染色体、起始位点、终止位点。value则对应了其他数据列。
# 和弦图示例
代码如下:
library(circlize)
genome=read.cytoband(species = hg19) # circlize包预制的hg19基因组
chr=data.frame(chr=names(genome$chr.len),start=1,end=genome$chr.len)
set.seed(12345) bed=generateRandomBed(nr = 50,species = "hg19") # 生成随机bed注释
bed$color=sample(rainbow(7),size = 55,replace = TRUE)
link1=generateRandomBed(nr=100)
link1$color=skyblue
link2=generateRandomBed(nr=100)
link2=link2[sample(1:nrow(link2), nrow(link2)),]
####设置输出文件####
# pdf(random_circle1.pdf)
circos.clear()
####和弦图初始化####
circos.initialize(factors = as.factor(a$chromosome),xlim = chr[,2:3])
####添加坐标轴####
circos.genomicTrackPlotRegion(chr, ylim = c(0, 1), bg.border = NA, track.height=0.05,panel.fun = function(region,value,...){
sector.index = get.cell.meta.data(sector.index)
track.index = get.cell.meta.data(track.index)
xlim = get.cell.meta.data(xlim, sector.index = sector.index, track.index = track.index)
major.by = 40000000
major.at = seq(floor(xlim[1]/major.by) * major.by, xlim[2], by = major.by)
major.at = c(major.at, major.at[length(major.at)] + major.by)
major.tick.labels=major.at/1000000
circos.axis(h = 1,labels.cex = 0.8, major.at = major.at, labels = major.tick.labels,labels.font=2,col="grey",minor.ticks = 0, sector.index = sector.index,track.index = track.index,major.tick.length = convert_y(1.5,mm,sector.index,track.index),lwd = 2)
circos.rect(xlim[1],0,xlim[2],1,col=grey,border=NA)
})
####添加基因块####
circos.genomicTrackPlotRegion(bed,ylim = c(0, 1), bg.border = NA, track.height=0.05,panel.fun = function(region,value,...){
sector.index = get.cell.meta.data(sector.index);print(sector.index)
track.index = get.cell.meta.data(track.index);print(track.index)
circos.rect(region$start,0,region$end,1,col=value$color,sector.index = sector.index,track.index = track.index,border=NA)
})
####添加link####
circos.genomicLink(link1, link2, col = link1$color, border = NA)
#dev.off()
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
# 结果图
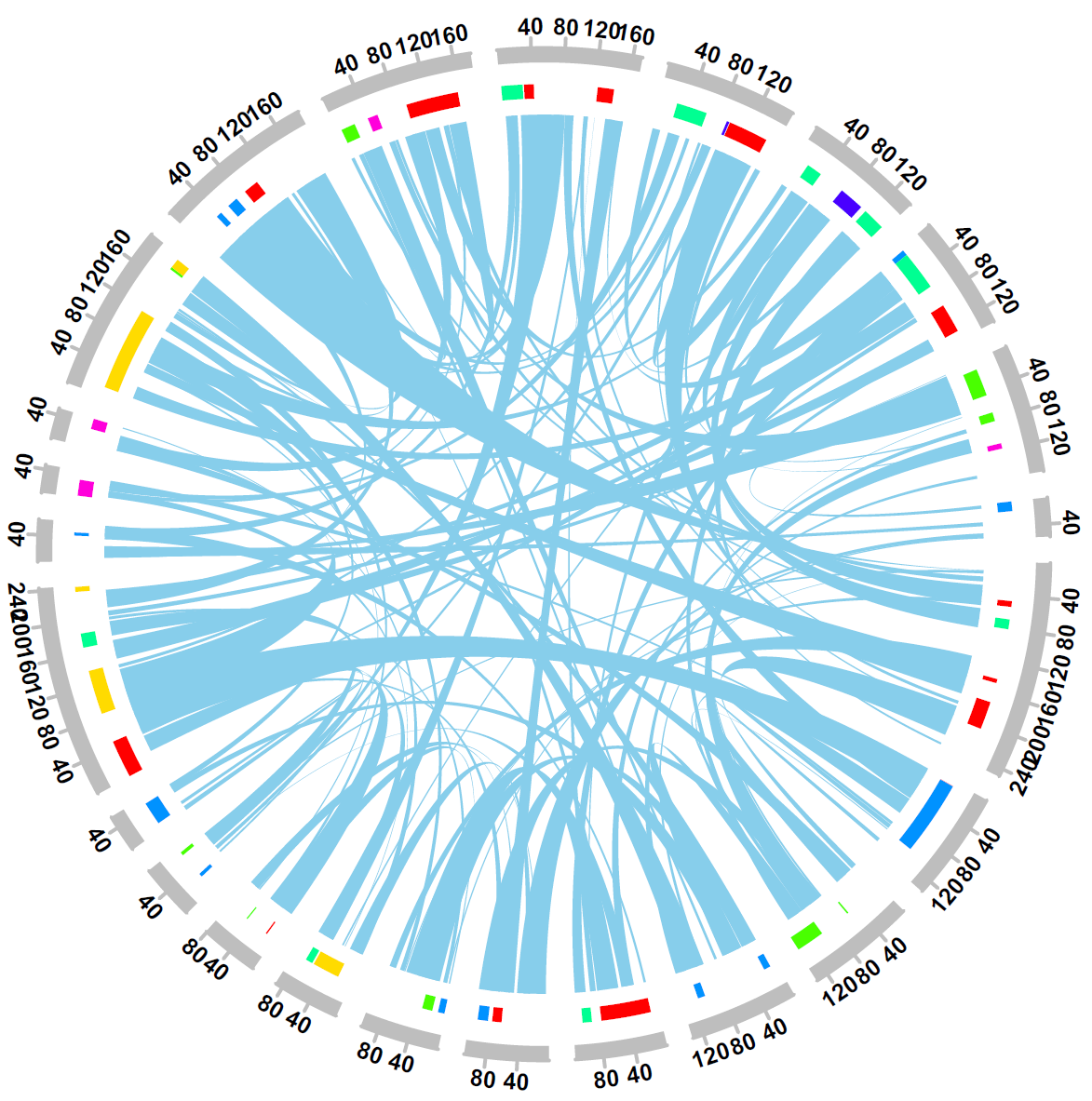 参考链接https://jokergoo.github.io/circlize_book/book/ (opens new window)
参考链接https://jokergoo.github.io/circlize_book/book/ (opens new window)
上次更新: 2024/03/11, 23:50:27